C . Baghdali, N S . Fedala.
Service d’Endocrinologie et de Maladies Métaboliques. C.H.U Bab El Oued
Le syndrome de Cushing d’origine endogène est une pathologie rare, due dans 80% des cas à un adénome hypophysaire à ACTH, plus rarement à un adénome surrénalien, un corticosurrénalome ou une sécrétion d’ACTH ectopique d’origine paranéoplasique. Les patients en hypercorticisme présentent des caractéristiques cliniques et biologiques proches de celles du syndrome métabolique
Les complications cardiovasculaires du syndrome de Cushing en font toute la gravité, avec un taux de mortalité 4 fois plus élevé chez les patients comparés à des sujets normaux. Ces complications sont dues notamment à une forte prévalence de facteurs de risque cardiovasculaires, à savoir une hypertension artérielle, une obésité abdominale, une insulino-résistance voire un diabète, une dyslipidémie et des anomalies de la viscosité sanguine .L’ensemble de ces anomalies joue un rôle dans l’augmentation du risque cardiovasculaire et de la morbi-mortalité non seulement au moment de la phase active de la maladie, mais également après guérison de l’hypercorticisme. Ainsi la prise en charge du patient porteur d’un hypercorticisme doit être multidisciplinaire, et une nouvelle évaluation des complications doit être maintenue au cours du suivi.
Introduction -Définition
Le syndrome de Cushing regroupe l’ensemble des symptômes cliniques et biologiques secondaires à un excès chronique de sécrétion de glucocorticoïdes par les glandes surrénales. Bien que ce syndrome soit rare, il demeure grave de par son évolution et son pronostic greffés par de multiples complications viscérales et métaboliques responsables d’une morbi- mortalité élevée [1].
Epidémiologie
Le syndrome de cushing a une incidence de 02–5/1 000000 et une prévalence de 39– 79/1 000000.
Le diagnostic est généralement posé à un âge moyen, la médiane étant de 40 ans; avec une nette prédominance féminine [2].
Diagnostic positif
Signes cliniques
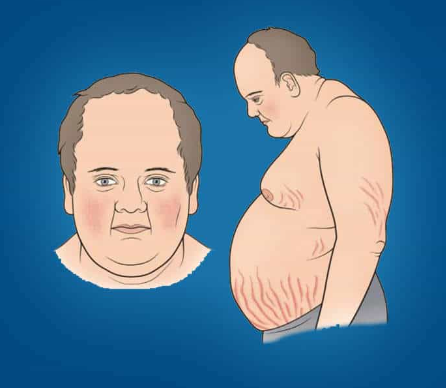
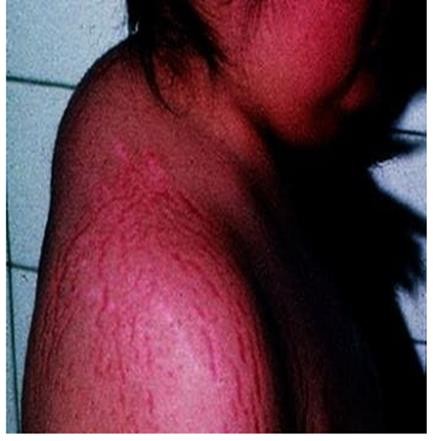
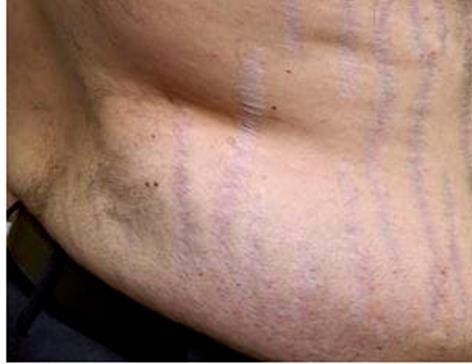
L’existence d’une prise de poids et d’une répartition facio-tronculaire de la masse grasse, d’une érythrose faciale et de signes d’hyper-catabolisme cutané, osseux ou musculaire (vergetures larges et pourpres, ostéoporose, amyotrophie…) sont évocateurs. Les troubles du cycle sont fréquents ainsi que l’hypogonadisme. Les troubles thymiques et l’asthénie peuvent aussi être présents.
Les symptômes cutanés et musculaires, reflets de l’activité catabolique et anti- anabolique du cortisol, sont les plus spécifiques et leur présence renforce considérablement la probabilité de syndrome de Cushing [2,3].
En pratique, ce diagnostic doit être évoqué devant un ensemble de symptômes associant une obésité androïde avec une redistribution facio-tronculaire des graisses, une hypertension artérielle et un diabète. Ce tableau recouvre celui du syndrome métabolique qui touche plus de 10 % des adultes en France.

Signes biologiques
En cas de suspicion de syndrome de Cushing, la première étape du diagnostic biologique a pour objectifs d’affirmer l’hypercorticisme et d’écarter les patients présentant un pseudo-Cushing.
Les examens utilisés sont :
- L’étude du cycle nycthéméral de la sécrétion du cortisol
- Le dosage du cortisol libre urinaire
- Les tests de freinage à la dexaméthasone
La cortisolémie matinale est peu discriminante du fait de larges chevauchements entre les valeurs normales et celles rencontrées dans le syndrome de Cushing. La perte du rythme circadien de la sécrétion du cortisol est un signe précoce du syndrome de Cushing. Mais la réalisation des prélèvements vespéraux est difficilement réalisable en ambulatoire et les ponctions veineuses soumettent le patient à un stress susceptible d’augmenter la cortisolémie.
Ces difficultés pratiques expliquent l’intérêt du dosage du cortisol salivaire. La salive est dépourvue des protéines de transport et le cortisol salivaire est étroitement corrélé au cortisol libre plasmatique. Le recueil de l’échantillon, non stressant, est facilement réalisable en ambulatoire mais ce dosage n’est pas disponible dans tous les laboratoires.
La cortisolurie des 24 heures est augmentée en cas d’hyperproduction de cortisol. Son dosage se heurte à la difficulté d’obtention d’un recueil urinaire correct et la mesure simultanée de la créatininurie permet d’en estimer la qualité. De plus, les fluctuations spontanées de la sécrétion dans l’hypercorticisme rendent impératif de recueillir les urines pendant 2 voire 3 journées consécutives.
La perte du rétrocontrôle physiologique des corticoïdes sur la sécrétion surrénalienne est étudiée grâce à la dexaméthasone (DMZ). Ce glucocorticoïde de synthèse se lie aux récepteurs du cortisol au niveau hypophysaire et supprime la libération d’ACTH chez l’individu sain. Les protocoles utilisés sont le freinage minute (le dosage du cortisol plasmatique ou salivaire matinal après la prise de 1 mg de DMZ la veille vers 23 h), réalisable en ambulatoire, et le freinage «faible » de Liddle ou freinage standard (0,5 mg de DMZ sont administrés toutes les six heures pendant 48 heures, la première prise débutant à 9 h. La mesure de la cortisolémie est réalisée six heures après la dernière prise de DMZ) qui permet de poser le diagnostic d’hypercorticisme endogène.
Diagnostic étiologique
Une fois le diagnostic de syndrome de Cushing posé, la première étape du diagnostic étiologique vise à établir l’ACTH dépendance de l’hypercorticisme par un dosage de l’ACTH plasmatique.
Une concentration inférieure à 5 pg/mL signe l’ACTH-indépendance, une concentration supérieure à 15 pg/mL signe l’ACTH-dépendance [2, 3].
Syndrome de cushing ACTH- dépendants: 80% des cas
- Maladie de Cushing
- Tumeurs ectopiques sécrétant l’ACTH
Syndrome de cushing ACTH – Indépendants : 20% des cas
- Adénome cortisolique bénin
- Corticosurrénalome
- Hyperplasie macronodulaire des surrénale
- Dysplasie micronodulaire bilatérale
- Maladie de McCune-Albrigt
Anomalies cardio-métaboliques au cours du syndrome de cushing
Atteintes cardiovasculaires
Elles jouent un rôle majeur dans l’augmentation de la mortalité associée au syndrome de Cushing.
L’HTA
L’HTA est présente chez environ 70 % des patients, l’amélioration de la pression artérielle et la diminution des besoins en traitement antihypertenseur sont observées chez la majorité des patients après guérison mais l’HTA peut persister chez environ 25% d’entre eux.
Sa persistance est attribuée à un remodelage des micro-vaisseaux artériels ou à la rémanence de l’adiposité abdominale et de l’insulino-résistance.
La durée de l’exposition à l’excès de cortisol est un facteur déterminant de la rémanence de l’HTA et constitue un facteur prédictif de surmortalité.
Sa pathogénie est complexe et fait intervenir un effet propre du cortisol qui sensibilise le muscle vasculaire aux agents vasoconstricteurs et inhibe la synthèse de monoxyde d’azote (NO), principal médiateur de la vasorelaxation dépendante de l’endothélium.
En cas d’hypercorticisme intense, les capacités d’inactivation du cortisol par l’enzyme 11 b hydroxy- steroïde – déshydrogénase rénale qui métabolise le
cortisol en cortisone peuvent être dépassées.
Dans ce cas, le cortisol peut se lier et activer le récepteur rénal des minéralo- corticoïdes, entraînant un syndrome d’excès apparent de minéralocorticoïdes [4].
Anomalies vasculaires
Une action spécifique des corticoïdes sur la paroi vasculaire est également suspectée.
Les perturbations de la crase sanguine concourent à l’augmentation du risque thrombotique en particulier dans les hypercortisolismes intenses [5].
Elles associent à la fois une hypercoagulabilité et une inhibition du système fibrinolytique [6].
L’ensemble de ces anomalies aboutit à une augmentation de la prévalence de l’atteinte athéro-sclérotique carotidienne et coronarienne.
Cette atteinte ne semble régresser que partiellement après la cessation de l’hypercorticisme.
Anomalies cardiaques
L’hypercortisolisme peut être à l’origine d’une atteinte structurelle et fonctionnelle du muscle cardiaque pouvant aboutir à une fibrose myocardique: hypertrophie, remodelage concentrique, dysfonctions systoliques, diastoliques, à l’origine dans certains cas à des cardiomyopathies dilatées (CMD) cortisoliques réversibles ou non après normalisation du cortisol.
Troubles métaboliques au cours du syndrome de cushing
- Troubles du métabolisme glucidique
L’intolérance aux hydrates de carbone et le diabète sucré patent sont rencontrés chez respectivement environ 60 et 20 % des patients et jouent un rôle majeur dans le risque cardiovasculaire associé au syndrome de Cushing [7].
Ce pourcentage est souvent sous-estimé car fondé sur la mesure de la glycémie à jeun. Lorsqu’une hyperglycémie provoquée par voie orale (HGPO) est réalisée, la prévalence de diabète sucré intéresse plus de 50 % des patients.
En effet, l’excès de cortisol favorise l’accrétion viscérale de tissu adipeux à l’origine de l’insulino-résistance.
Par ailleurs, le cortisol stimule la néoglucogenèse en activant la synthèse d’enzymes hépatiques-clés (PEPCK), en augmentant l’afflux hépatique de substrats néoglucogéniques via ses effets protéo- et lipolytiques et en exerçant un effet permissif vis-à-vis de l’action périphérique du glucagon et des cathécolamines [8].
Enfin, les glucocorticoïdes inhibent la captation périphérique du glucose par son transporteur GLUT-4.
Le diagnostic de syndrome de cushing doit être évoqué devant un diabète de type 2 déséquilibré sans cause évidente avec une surcharge pondérale, de morphotype androïde, une HTA résistante aux traitements chez un sujet relativement jeune.
- Troubles du métabolisme lipidique
Les troubles de la tolérance aux hydrates de carbone sont généralement associés à des anomalies lipidiques caractéristiques
de l’insulino-résistance chez plus de 50 % des patients : hypertriglycéridémie, diminution du high density lipoprotein (HDL)-cholestérol et, à un moindre degré, augmentation du low density lipoprotein (LDL)-cholestérol [8].
Ces anomalies métaboliques s’améliorent en général après rémission de l’hypercortisolisme, mais la prévalence du diabète sucré et des dyslipidémies demeure accru par rapport à une population de sujets appariés sur l’âge et l’indice de masse corporelle (IMC).
Chez ces patients, la tolérance aux hydrates de carbone est inversement corrélée au tour de taille, suggérant que la persistance d’anomalies de la distribution des graisses joue un rôle physiopathologique clé.
Quelques études suggèrent que la persistance de ces facteurs de risque cardiovasculaire est corrélée à la durée de l’hypercortisolisme.
En effet les glucocorticoides induisent une augmentation de la lipolyse périphérique, favorisent la libération des acides gras dans le sang, leur captation par le foie et donc leur accumulation dans celui-ci.
- Retentissment de l’hypercorticisme sur la fonction hépatique
Stéatose hépatique :
Les glucocorticoides induisent une augmentation de la lipolyse périphérique, et favorisent la libération des acides gras dans le sang, leur captation par le foie et donc leur accumulation dans celui-ci.
On observe une vacuolisation des hépatocytes (augmentation de la taille et du
nombre des vacuoles), une accumulation de glycogène et de lipides dans le cytoplasme, et une baisse du nombre de mitochondries.
Les phosphatases alcalines (PAL)
Dans l’hypercorticisme, les PAL qui sont des marqueurs de cholestase sont fortement augmentées dans 79 à 90% des cas, mais aucun signe histologique de cholestase n’est observé. L’augmentation des PAL est due à une augmentation de l’induction enzymatique cortico-induite.
L’alanine aminotransférase (ALAT)
L’ALAT qui est un marqueur de cytolyse est qui fortement augmenté dans l’hypercorticisme, dans 50 à 74% des cas. Or, il n’y a pas, dans l’hyperorticisme, de signe histologique de nécrose cellulaire.
Par contre, l’ALAT est une enzyme importante de la gluconéogenèse. Celle-ci étant augmentée dans l’hypercorticisme, il y a une augmentation de la production d’ALAT.
γGlutamyl transférase (γGT)
Les γGT se trouvent dans le pancréas, le foie, le rein. Elles se trouvent dans le cytosol associées aux membranes cellulaires près des canaux biliaires. C’est donc un indicateur de cholestase.
Dans l’hypercorticisme, on constate une augmentation des γGT mais dans des proportions moindres que l’ALAT et les PAL.
Les glucorticoïdes induiraient une augmentation de leur synthèse hépatique.
- Troubles ioniques
Une hypokaliémie peut se rencontrer au cours du syndrome de cushing, sa sévérité est proportionnelle à la sévérité du syndrome de cushing, celle-ci s’explique par le mécanisme physiopathologique suivant :
Au niveau rénal, le récepteur minéralo- corticoïde possède la même affinité pour l’aldostérone et le cortisol.
La sélectivité de la réponse biologique est assurée par la présence d’une enzyme : la 11b-hydroxystéroïde deshydrogénase de type2 (11b-HSD 2).
Celle-ci métabolise le cortisol en cortisone présentant une faible affinité pour le récepteur minéralo-corticoïde qui est alors protégé d’une occupation« illicite » par le glucocorticoïde.
Cependant, la 11b-HSD 2 est saturée pour des concentrations proches des valeurs physiologiques supérieures du cortisol. Dès lors, en cas d’hypercorticisme, l’enzyme ne peut métaboliser tout le cortisol. Le glucocorticoïde en excès se lie au récepteur minéralo-corticoïde et mime l’action de l’aldostérone à l’origine de l’alcalose avec déplétion potassique [8]; entraînant un syndrome d’excès apparent de minéralo- corticoïde [4]. L’hypokaliémie par fuite urinaire du potassium peut aussi être secondaire à l’hypersécrétion de cortisol, indépendamment d’un
hyperaldostéronisme. Une alcalose hypokaliémique avec kaliurèse conservée ou augmentée oriente vers une sécrétion ectopique d’ACTH ou un carcinome surrénalien.
Traitement
Les manifestations du syndrome de Cushing sont au moins partiellement, voire totalement, réversibles lorsque l’excès de sécrétion de cortisol est aboli.
La disparition des anomalies morphologiques est quelquefois très impressionnante et les patients sont véritablement « transformés », reprenant leur apparence normale en quelques mois.
De la même façon, les autres manifestations comme le diabète, l’hypertension artérielle, les troubles psychiques, menstruels sont corrigés ou nettement améliorées.
Certaines manifestations de l’hypersécrétion de cortisol régressent de façon moins spectaculaire : des vergetures importantes peuvent laisser des cicatrices durables, et lorsque l’ostéoporose a entraîné des tassements vertébraux, la perte de taille est irréversible.
D’une façon générale, les manifestations d’un syndrome de Cushing régressent d’autant mieux qu’elles sont modestes, récentes, et que le patient est jeune.
La chirurgie surrénalienne dans le syndrome de Cushing, qu’elle soit unilatérale ou bilatérale, est le plus souvent réalisée par laparoscopie qui permet un abord trans- ou rétropéritonéal, minimisant les problèmes de cicatrisation postopératoire qui sont importants dans un contexte d’imprégnation cortisolique. Pour des raisons carcinologiques, un abord large à ciel ouvert, permettant une résection en bloc de la tumeur, du rein et des organes adjacents, est réalisé en cas de corticosurrénalome malin.
Dans la maladie de Cushing l’exérèse chirurgicale des microadénomes hypophysaire par voie transphénoidale est le traitement de choix. Le succès et la morbidité de la chirurgie dépendent étroitement de l’expérience du chirurgien.
Conclusion
La fréquence des complications cardiovasculaires et métaboliques du syndrome de Cushing en font toute la gravité, avec un taux de mortalité 4 fois plus élevé chez les patients comparés à des sujets normaux. Le traitement du syndrome de cushing bien que délicat dans certains cas reste indispensable.
Références bibliographiques
- Arnaldi G, Angeli A, Atkinson AB, Bertagna X, Cavagnini F, Chrousos GP, Fava GA, Findling JW, Gaillard RC, Grossman AB, Kola B, Lacroix A, Mancini T, Mantero F, Newell-Price J, Nieman LK, Sonino N, Vance ML, Giustina A, Boscaro M. Diagnosis and complications of Cushing’s syndrome : a consensus statement. J Clin EndocrinolMetab. 2003 ;88:5593-602.
- Newell-Price J, Bertagna X, Grossman AB, Nieman LK.Cushing’s syndrome. Lancet 2006 ; 367 : 1605-17.
- Tabarin A, Collet D, San Galli F, Paire JP, Loiseau H. Syndrome de Cushing. Paris : Elsevier, 2006, EMC Endocrinologie-Nutrition: 10-015-B- 10.
- Walker BR, Campbell JC, Fraser R, Stewart PM, Edwards CR. Mineralocorticoid excess and inhibition of 11 beta-hydroxysteroiddehydrogenase in patients with ectopic ACTH syndrome. ClinEndocrinol (Oxf) 1992;37:483-92.
- Findling JW, Raff H,Cushing’ssyndrome:important issues in diagnosis et management.J ClinEndocrinolMetab2006;91:3746-53.
- Bourdeau I, Bard C, Forget H, Boulanger Y, Cohen H, LacroixA.Cognitive function and cerebral assessment in patients who have Cushing’s syndrome. EndocrinolMetabClin North Am 2005;34: 357-69.
- Faggiano A, Pivonello R, Spiezia S, De Martino MC, Filippella M, DiSomma C, et al. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing’s disease during active disease and 1 year after disease remission. J ClinEndocrinolMetab2003;88:2527-33.
- Andrews RC, Walker BR. Glucocorticoids and insulin resistance:oldhormones, new targets. ClinSci1999;96:513-23.
- Quinkler M, Stewart PM. Hypertension and the cortisol-cortisonshuttle.JClinEndocrinolMetab2003; 88 : 2384-92